
长久以来,抗肿瘤治疗是学术界高度关注的热点问题。随着现代临床医学在遗传及分子科学领域的不断发展,越来越多的新兴抗肿瘤药物逐渐从基础研究走向临床实践。在表观遗传学领域,赖氨酸乙酰转移酶(KAT)抑制剂是当下新兴的一类靶向药物,具有抗肿瘤治疗价值和临床应用潜力。本文从表观遗传学机制入手,总结 KAT 类药物的作用机制,研发现状,代表药物及未来发展,以期为临床医生提供参考。
一. 表观遗传学与KAT抑制剂
- 1.表观遗传学的定义与相关机制
表观遗传学描述的内容为基因功能的可遗传变化,但这些变化并不涉及 DNA 碱基序列的改变。目前学者将表观遗传学定义为:通过 DNA 修饰、组蛋白修饰、RNA 修饰、染色质重塑和非编码 RNA 调节五个关键机制控制染色质状态调节系统(见图 1,2),这些机制可独立传递 DNA 碱基序列中的遗传信息,并可受某些生理/病理性影响而激活/抑制某些基因组区域的表达,在生物体发育和体内平衡中发挥重要作用[1]。
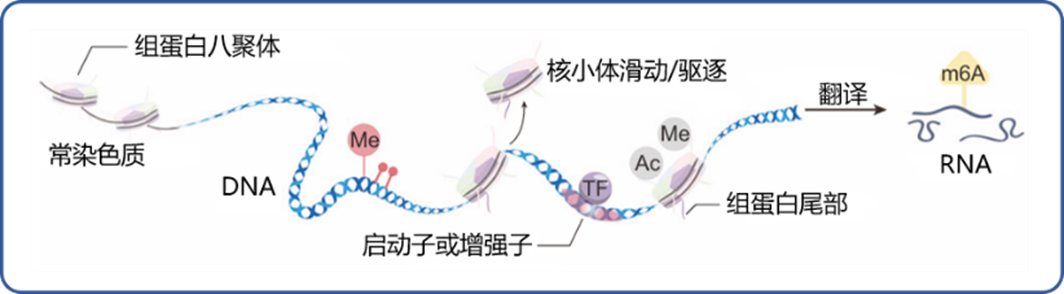
图片改编自文献:Dai W, Qiao X, Fang Y, et al. Epigenetics-targeted drugs: current paradigms and future challenges. Fig 1a
DNA:脱氧核糖核酸;Me:甲基;TF:转录因子;Ac:乙酰基;RNA:核糖核酸;m6A:N6-腺苷酸甲基化
图1 表观遗传修饰5大关键机制总结图例
简言之,表观遗传修饰是指通过对特定碱基或氨基酸进行修饰或擦除修饰,从而对基因表达产生各种影响。这些修饰的调节通常依赖各种酶类完成,而依照酶的功能不同又可分为写入因子,读取因子和擦除因子等类别。其中,写入因子参与修饰特定碱基或氨基酸,擦除因子参与修饰的清除,而读取因子则可识别修饰状态下特定遗传构象蛋白质并加以结合(见图 2)[1]。以组蛋白修饰为例,KAT——此前被称作组蛋白乙酰化转移酶(HAT)[2]可作为写入因子,在组蛋白特定构象上写入乙酰基修饰以使组蛋白乙酰化;这种乙酰基修饰也可通过擦除因子——即组蛋白相关去乙酰化酶来清除(见图 2)[1]。
在临床应用的可行性方面,研究者发现参与表观遗传修饰的各种酶之间存在着机制互补的特性,这一发现强调了表观遗传修饰存在可逆性。研究者进而利用这种特性,开发了以靶向手段调节表观遗传修饰的酶类,从而达到调控基因表达的治疗策略[1]。这一策略也为抗肿瘤治疗提供了新的思路。
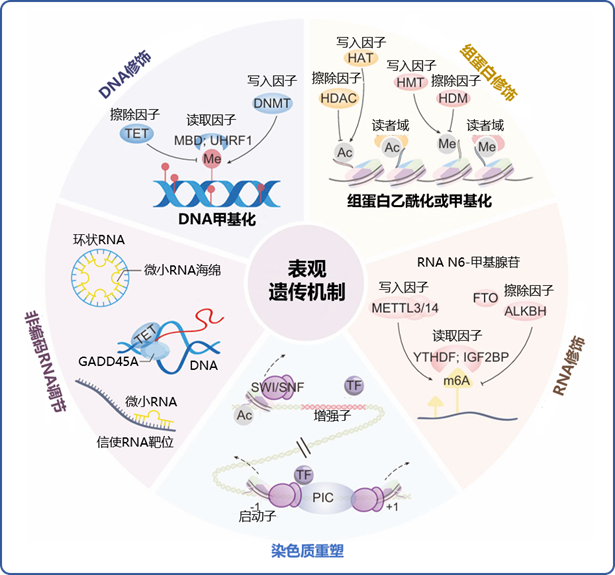
图片改编自文献:Dai W, Qiao X, Fang Y, et al. Epigenetics-targeted drugs: current paradigms and future challenges. Fig 1b
DNA:脱氧核糖核酸;RNA:核糖核酸;TET:十-十一易位修饰蛋白;MBD:甲基化 CpG 结合域蛋白;UHRF1:泛素样含植物同源结构域和环指结构域 1;DNA 甲基转移酶;HDAC:组蛋白去乙酰化酶;HAT:组蛋白乙酰化转移酶;HMT:组蛋白甲基化转移酶;HDM:组蛋白去甲基化酶;GADD45A:生长停滞和 DNA 损伤诱导的α基因;SWI/SNF:哺乳动物转换/蔗糖不发酵染色质重塑因子;PIC:转录起始复合物;METTL3/14:一种 m6A 甲基化转移酶;FTO:脂肪量和肥胖相关蛋白;ALKBH:RNA 去甲基化酶 AlkB 同源物;YTHDF:YT521-B 同源结构域家族蛋白; IGF2BP:胰岛素样生长因子 2 mRNA 结合蛋白
图2 表观遗传修饰各关键机制中的相关酶促反应
- 2. KAT 抑制剂的作用机制及作为抗肿瘤治疗靶向药物的理论基础
作为参与基因表达调节的表观遗传修饰酶类,KAT 家族包含利用乙酰辅酶A等在赖氨酸残基处翻译后修饰组蛋白/非组蛋白的不同酶,这些酶有助于诱导染色质松散,进而在蛋白质访问基因组并执行其功能(DNA 转录、复制或修复)方面发挥关键作用[3]。KAT 家族可分为多个子类,包括 MYST(Moz、Ybf2/Sas3、Sas2、Tip60)家族,p300 和环磷腺苷效应元件结合蛋白(p300/CBP)家族等。而在每个子类下又包含多种不同催化结构域的酶,如 MYST 家族中包含的 KAT6A、KAT6B 等,p300/CBP 家族中包含 KAT3A(CREBBP)和 KAT3B(EP300)等 [2,3]。
经组蛋白乙酰化修饰后的染色质可通过特异性蛋白质结构域读取,而溴结构域是其中重要一类[3]。该结构域由 4 个反向平行的 α 螺旋 αZ、αA、αB 和 αC 构成,并形成 ZA 环和 BC 环,两个环中的氨基酸残基形成一个疏水空腔,可识别并以氢键形式结合乙酰化赖氨酸残基以调控相关功能[4](见图 3 示例)[5]。
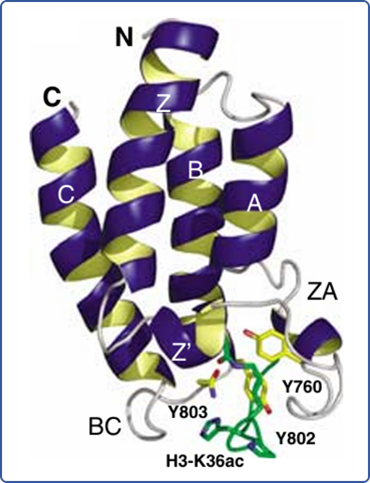
图片改编自文献:Structure and acetyl-lysine recognition of the bromodomain. Oncogene. Fig1a
黑字 C 和 N:碳端和氮端;白字 C/Z/B/A/Z’:溴结构域亚基αC/Z/B/A/Z’;H3-K36ac:组蛋白 H3 亚基 36 位赖氨酸乙酰化;Y760/802:酪氨酸残基;Y803:天冬氨酸残基
图3 乙酰化修饰组蛋白与溴结构域结合三维示意图
既往研究发现,KAT 的异常与恶性肿瘤间存在内在联系。其一,某些 KAT(如 KAT6A 等)相关复合物可能在乳腺癌等恶性肿瘤患者中表达扩增。其二,KAT 分子层面的构象改变可在总体上影响组蛋白乙酰化,进而在基因层面引发致癌基因或肿瘤抑制基因转录的特异性改变,这种分子改变常见于 KAT 相关复合物(如 EP300/CREBBP、KAT6A/KAT6B)的旁系同源酶亚基中(例如恶性肿瘤患者的EP300/CREBBP致死性合成突变)。其三,KAT 可作为一些重要细胞信号转导和细胞周期通路的转录共激活因子,通过相互作用以及致癌转录因子乙酰化调节机制,参与恶性肿瘤的发生发展[3]。此外,一些溴结构域通过识别和结合组蛋白乙酰化赖氨酸,招募染色质调控因子,促进基因转录,也可促进肿瘤的发生发展和转移[4]。
相关基因测序数据表明,在一些恶性肿瘤中存在 KAT 活性增加或功能丧失,这体现了恶性肿瘤具有 KAT 依赖性(图 4)。这种依赖性使得 KAT 成为一类有前景的新兴抗肿瘤药物靶点。且目前已有多种相关在研药物(图 4)[3]。
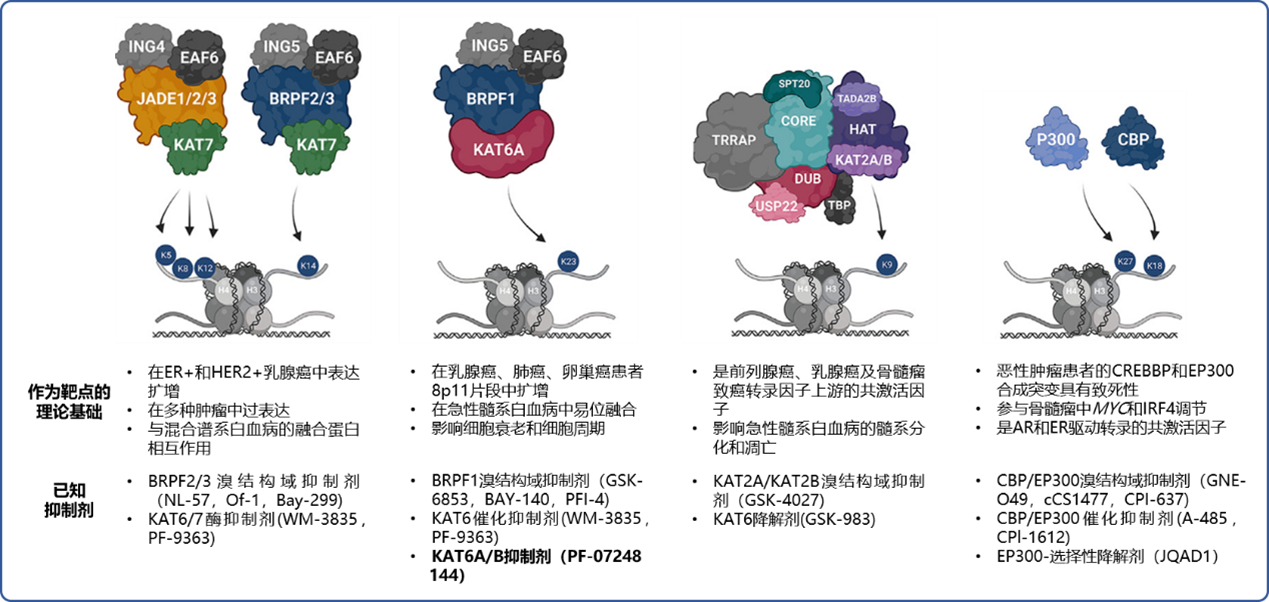
图片改编自文献:White J, Derheimer FA, Jensen-Pergakes K, et al. Histone lysine acetyltransferase inhibitors: an emerging class of drugs for cancer therapy. Fig 2
ING:生长抑制因子家族成员;EAF6:乙酰转移酶 NuA4 复合体之一;JADE:翡翠家族植物同源结构域指;KAT:赖氨酸乙酰转移酶;BRPF:含溴结构域和植物同源结构域指;TRRAP:转录域相关蛋白;SPT20:Spt-Ada-Gcn5-乙酰转移酶核心模块之一;DUB:去泛素化酶;TADA2B:转录衔接子 2B;HAT:组蛋白乙酰转移酶;TPB:TATA 结合蛋白;CBP/P300:同源蛋白,属蛋白质赖氨酸乙酰转移酶及共激活剂,该家族中包括 CREBBP 和EP300;ER:雌激素受体:HER:人表皮生长因子受体;8p11:染色体片段;MYC:骨髓瘤相关中心致癌基因;IRF4:干扰素调节因子 4;AR:雄激素受体
图4 KAT 作为肿瘤学靶点的理论基础和已知抑制剂
二. KAT 抑制剂的研发现状与代表药物
- 1. KAT 抑制剂的研发现状
赖氨酸乙酰化作为组蛋白及其它蛋白质翻译后的主要修饰过程,需通过 KAT 催化,并由其介导与溴结构域和其它读取因子相互作用[6],故而 KAT 催化结构域与相关溴结构域是研究者重点关注的治疗靶点(见表1)。
表1 目前 KAT 抑制剂的临床研发情况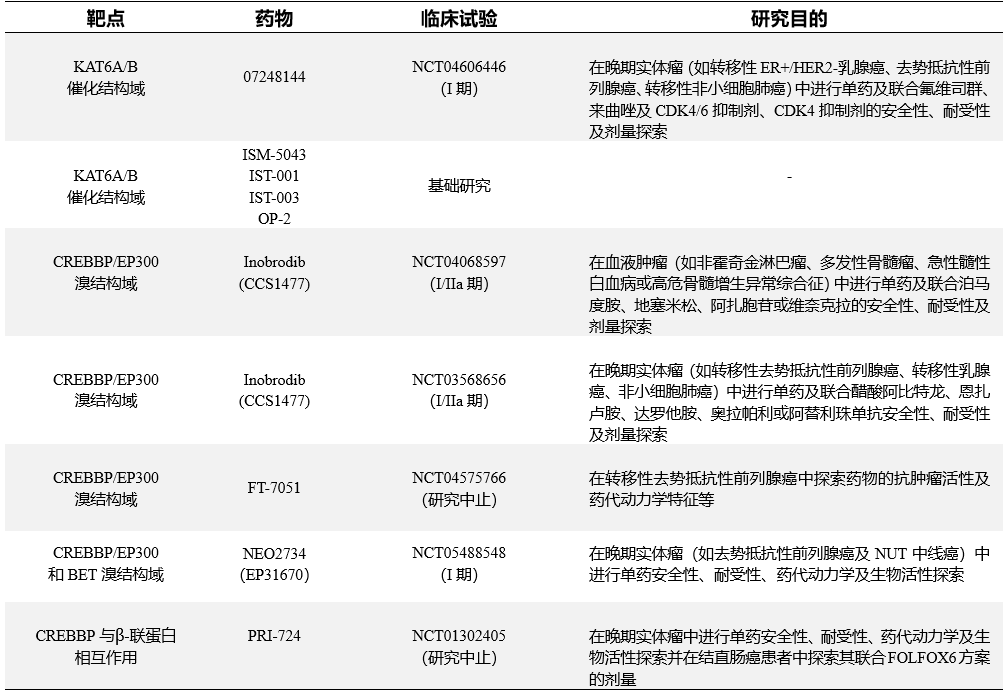
BET:含溴结构域和额外终端域家族蛋白; NUT 中线癌:伴有睾丸核蛋白(Nuclear protein of the testis, NUT) 基因 t(15;19) (q13; p13.1) 易位的未分化癌或中线致死性癌
既往研究者曾通过抑制乙酰化赖氨酸相关溴结构域的读取因子来抑制乙酰化途径,并研发出如含溴结构域和额外终端域家族蛋白(BET)抑制剂等药物,此后又转向开展了靶向 KAT 小分子抑制剂的研发。在靶向 KAT 研发早期,研究者尝试通过找到 KAT 蛋白或其亚基中溴结构域的小分子配体,以抑制溴结构域-配体蛋白相互作用的方式影响 KAT 功能,但结果证实其不能清除相关表性基因或达到酶抑制[3]。
此后,研究者以具有氨基酸差异的特异性催化结构域和溴结构域为靶点,研发了新一代 KAT 抑制剂,此类药物靶点的特异性保证了药物在KAT家族中的高选择性。在最近的研究中,靶向 KAT6A/KAT6B 催化结构域等多种新型KAT抑制剂取得研究进展,并在雌激素受体阳性(ER+)且人表皮生长因子受体 2 阴性(HER2-)乳腺癌等多种恶性肿瘤治疗中表现出临床应用潜力(见表 1) [3]。
- 2. KAT 抑制剂的临床疗效
在一项 I 期临床研究中 KAT6 抑制剂被证实具有显著的抗肿瘤作用。该研究纳入细胞周期蛋白依赖性激酶 4/6(CDK4/6)抑制剂和内分泌治疗失败后的 ER+HER2- 转移性乳腺癌患者,并给予 KAT6 抑制剂单药及联合氟维司群方案,研究旨在评估单药及联合方案的疗效和安全性[7]。
在单药治疗剂量扩展亚组(n=35)中,所有患者既往均接受内分泌及 CDK4/6 抑制剂治疗,接受化疗和氟维司群治疗的患者比例分别为 74.3% 和 80.0%,亚组研究显示出 KAT6 抑制剂具有抗肿瘤活性,在晚期重度经治(中位 5 线)的患者中,客观缓解率(ORR)为 11.4%(95% CI:3.2~26.7),中位无进展生存期(mPFS)为 3.3 个月(95% CI:2.0~5.8)[7]。
在KAT6抑制剂联合氟维司群方案的亚组(n=43)中,所有患者既往均接受内分泌及 CDK4/6 抑制剂治疗,接受化疗和氟维司群治疗患者的比例分别为 27.9%和 11.6%,在该亚组研究中,观察到患者 ORR 为 30.2%(95% CI:17.2~46.1),mPFS 为 10.7 个月(95% CI:5.3~不可评估)。而其中接受过三线及以上治疗的患者经联合方案治疗后 ORR 高达 40.0%(95% CI:19.1~63.9),且 mPFS 与该亚组总体分析结果相同,也为 10.7 个月(95% CI:5.5~不可评估)[7]。
此外,该研究的肿瘤样本和外周血单核细胞分析证实 KAT6 抑制剂还对 KAT6A/KAT6B 催化结构域具有显著的抑制作用。上述结果显示出 KAT6 抑制剂在既往曾接受多种治疗的 ER+HER2- 转移性乳腺癌患者中具有持久的抗肿瘤活性与良好的安全性,该结论确定了 KAT6A/KAT6B 催化结构域可作为恶性肿瘤的治疗靶点,也为转移性乳腺癌提供了新的治疗途径[7]。
三. KAT 抑制剂应用的未来展望
如前文所述,KAT 的分子异常与恶性肿瘤发生发展密切相关。在现有的理论研究支持下,以表观遗传学机制为基础的 KAT 抑制剂无疑在恶性肿瘤治疗相关临床应用中具有很高的潜力。在临床研究方面,以 KAT6A/KAT6B 催化结构域为靶点的新一代 KAT 抑制剂取得了一定积极成果。KAT6 抑制剂在 ER+HER2- 转移性乳腺癌具有显著的抗肿瘤活性和良好的安全性,有望在未来投入临床实践,为 CDK4/6 经治的晚期乳腺癌患者治疗提供新方案。
KAT 抑制剂在恶性肿瘤耐药及联合治疗方面同样颇具潜力。众所周知,转移性乳腺癌长期用药可能导致耐药问题,其中常见原因为 ESR1(编码 ER 本身的基因)突变[8]。而 KAT6A 是 ESR1 转录的直接调节因子,通过抑制 KAT6A/KAT6B,可从转录本层面抑制 ESR1 相关表达。在氟维司群耐药的乳腺癌模型中,KAT6 抑制剂也抗肿瘤活性[3]。这些结果显示 KAT 抑制剂在抵抗耐药,并与氟维司群联合抗肿瘤治疗方面具有应用价值。
总结
随着研究深入,越来越多的新兴抗肿瘤药物进入人们的视野。在表观遗传学层面,KAT 异常与恶性肿瘤的发生发展密切相关,靶向 KAT 治疗恶性肿瘤成为未来拥有广泛临床应用前景的新策略。目前,以 KAT6 抑制剂为代表的一系列靶向 KAT 药物在研,研究涉及乳腺癌、前列腺癌、肺癌等多个瘤种,并取得一定的积极成果,显示出此类药物在恶性肿瘤治疗中的临床应用潜力。总体而言,KAT 类药物的出现,为恶性肿瘤精准医学治疗的发展注入了新动力,也为包括乳腺癌在内的广大癌症患者带来新的希望。
参考文献
[1] Dai W, Qiao X, Fang Y, et al. Epigenetics-targeted drugs: current paradigms and future challenges. Signal Transduct Target Ther. 2024 Nov 26;9(1):332.
[2] Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. 2019 Mar;20(3):156-174.
[3] White J, Derheimer FA, Jensen-Pergakes K, et al. Histone lysine acetyltransferase inhibitors: an emerging class of drugs for cancer therapy. Trends Pharmacol Sci. 2024 Mar;45(3):243-254.
[4] 刘晓庆,梁爽,刘永军,等.溴结构域蛋白4的抑制策略及其在肿瘤治疗中的研究进展[J].中国药科大学学报,2021,52(03):270-278.
[5] Mujtaba S, Zeng L, Zhou MM. Structure and acetyl-lysine recognition of the bromodomain. Oncogene. 2007 Aug 13;26(37):5521-7.
[6] Zhou MM, Cole PA. Targeting lysine acetylation readers and writers. Nat Rev Drug Discov. 2024 Nov 21.
[7] Mukohara T, Park YH, Sommerhalder D, et al. Inhibition of lysine acetyltransferase KAT6 in ER+HER2- metastatic breast cancer: a phase 1 trial. Nat Med. 2024 Aug;30(8):2242-2250.
[8] Lloyd MR, Jhaveri K, Kalinsky K, et al. Precision therapeutics and emerging strategies for HR-positive metastatic breast cancer. Nat Rev Clin Oncol. 2024 Oct;21(10):743-761.
审批编码:PP-UNP-CHN-1076 有效期至:2027-3-20



