
靶向治疗是肿瘤治疗的重要手段,然而许多肿瘤相关靶点没有催化活性口袋的蛋白质,导致传统的占位驱动型抑制剂难以靶向结合[1]。蛋白水解靶向嵌合体(PROTAC)等药物运用靶向蛋白降解技术(TPD)的新兴手段打破了这一限制,扩展了可靶向的蛋白质种类[1]。在肿瘤领域,乳腺癌作为影响世界女性健康的常见恶性肿瘤,在全球范围内发病率持续上升[2],需要临床医生加以重视。近年来,PROTAC在治疗乳腺癌相关临床研究中取得了突破性成就,为乳腺癌治疗提供了新思路。本文总结PROTAC类药物的作用机制,临床前景以及其在乳腺癌治疗中的应用,以期为临床工作者提供参考。
一. PROTAC类药物及应用前景简介
1. PROTAC类药物设计的理论基础与作用机制
从生理学层面看,在精确的时间和部位维持蛋白质同质化和调节蛋白质水平,是生物体及细胞的重要机制[3]。在这一调节机制中,蛋白质的清除是重要环节。对哺乳动物而言,蛋白质清除主要受泛素蛋白酶体系统(UPS)和自噬-溶酶体途径控制,其中UPS负责大部分蛋白质降解相关的调节以及清除错误折叠或受损的蛋白质(见图1) [3]。
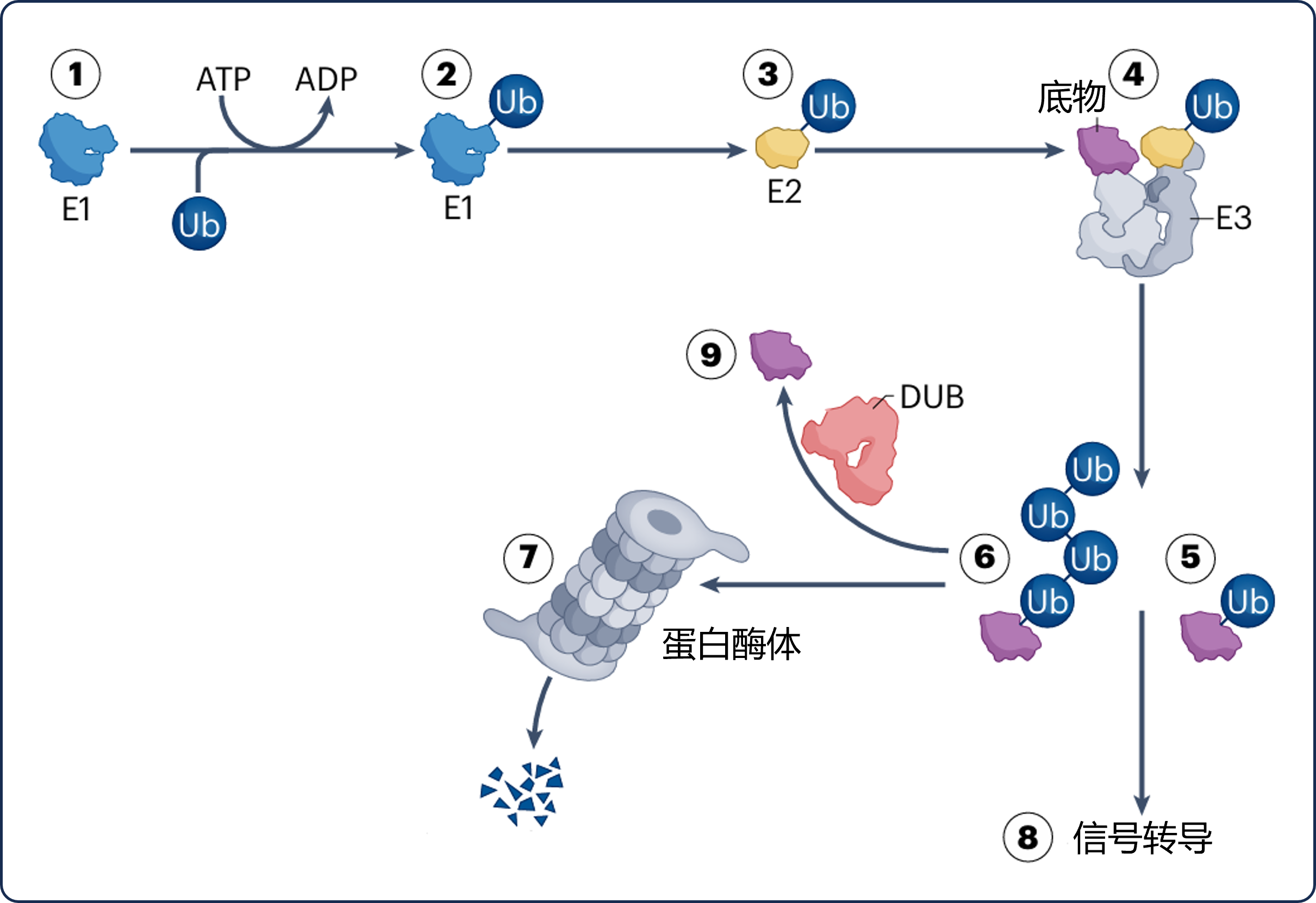
图1 经泛素蛋白酶体系统(UPS)降解蛋白质的机制
图片改编自:Targeted protein degradation: from mechanisms to clinic. Fig.1a
E1、E2、E3:泛素连接酶E1、E2、E3;ATP:三磷酸腺苷;ADP:二磷酸腺苷;Ub:蛋白质经泛素化反应修饰;DUB:去泛素化酶
TPD是利用小分子诱导特定靶蛋白降解的技术手段。最初,研究人员通过“将靶蛋白募集至泛素连接酶”的方法证实了这一理念,并在此后扩展应用至其它许多细胞降解途径中。目前,TPD通常指由UPS介导的蛋白降解,由于此途径需通过数百种不同的泛素连接酶E3识别靶蛋白来完成,因此具有特异性[3]。而PROTAC作为蕴含TPD设计理念的一类模块化分子,其可通过相应靶点结合靶蛋白与泛素连接酶E3,形成靶蛋白-PROTAC-连接酶构型的三元复合物(异双功能分子)(见图2)[3]。
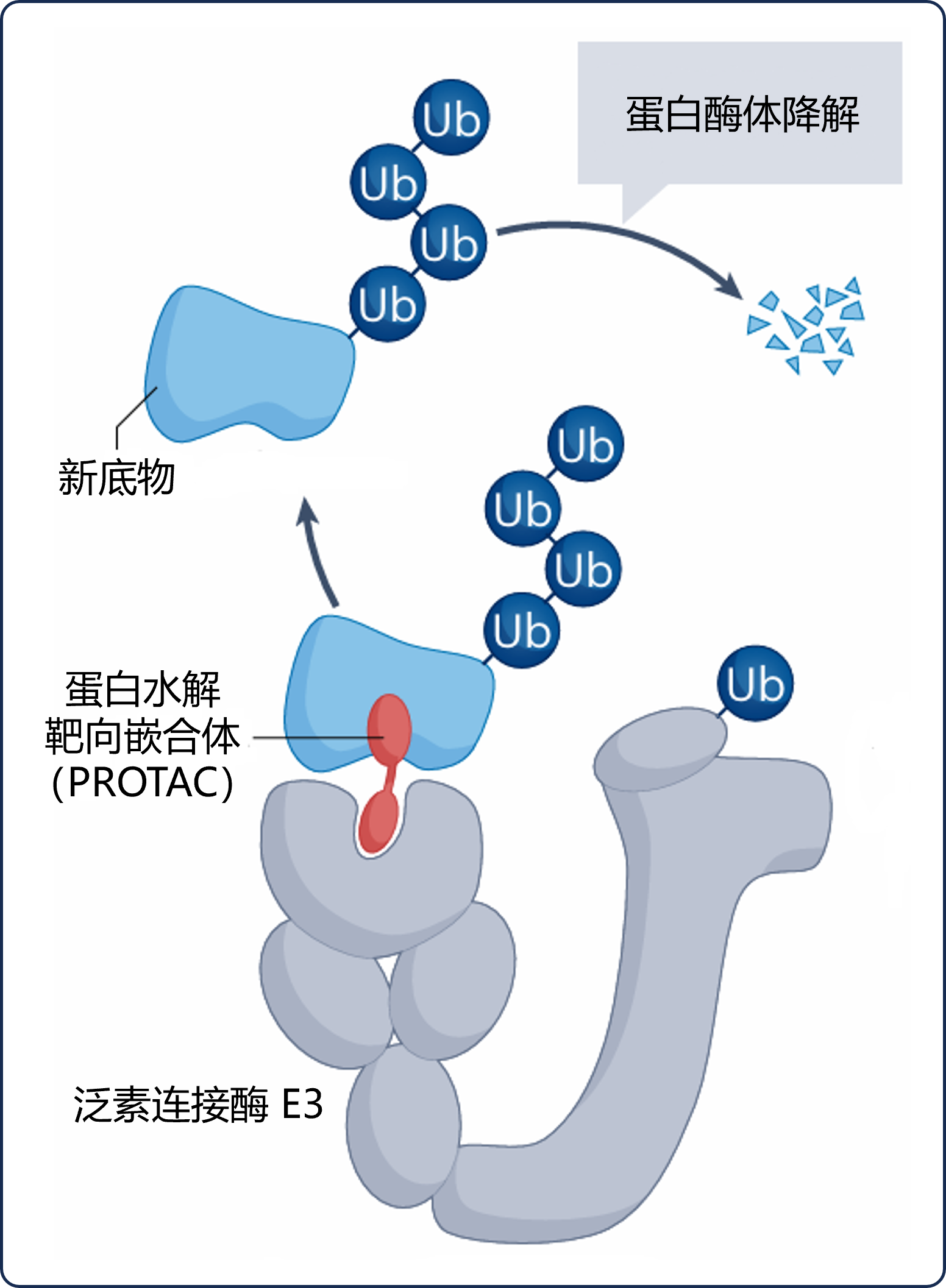
图2 靶蛋白-PROTAC-连接酶 三元复合物构象图
图片改编自:Targeted protein degradation: from mechanisms to clinic. Box-1(左图)
Ub:蛋白质经泛素化反应修饰
在具体作用机制上,PROTAC降解蛋白质涉及泛素连接酶E1、E2、E3三种酶促反应,使目标底物泛素化,泛素化目标底物可进一步被26S蛋白酶体识别,进而完成蛋白降解(见图3),利用这一机制有利于开展对恶性肿瘤的治疗。目前已发现600余种介导此过程的泛素连接酶E3,但已知具有小分子配体的目前仅有1%,其中最常用于研究的配体有Cereblon(CRBN)、Von Hippel-Lindau(VHL)等[2]。
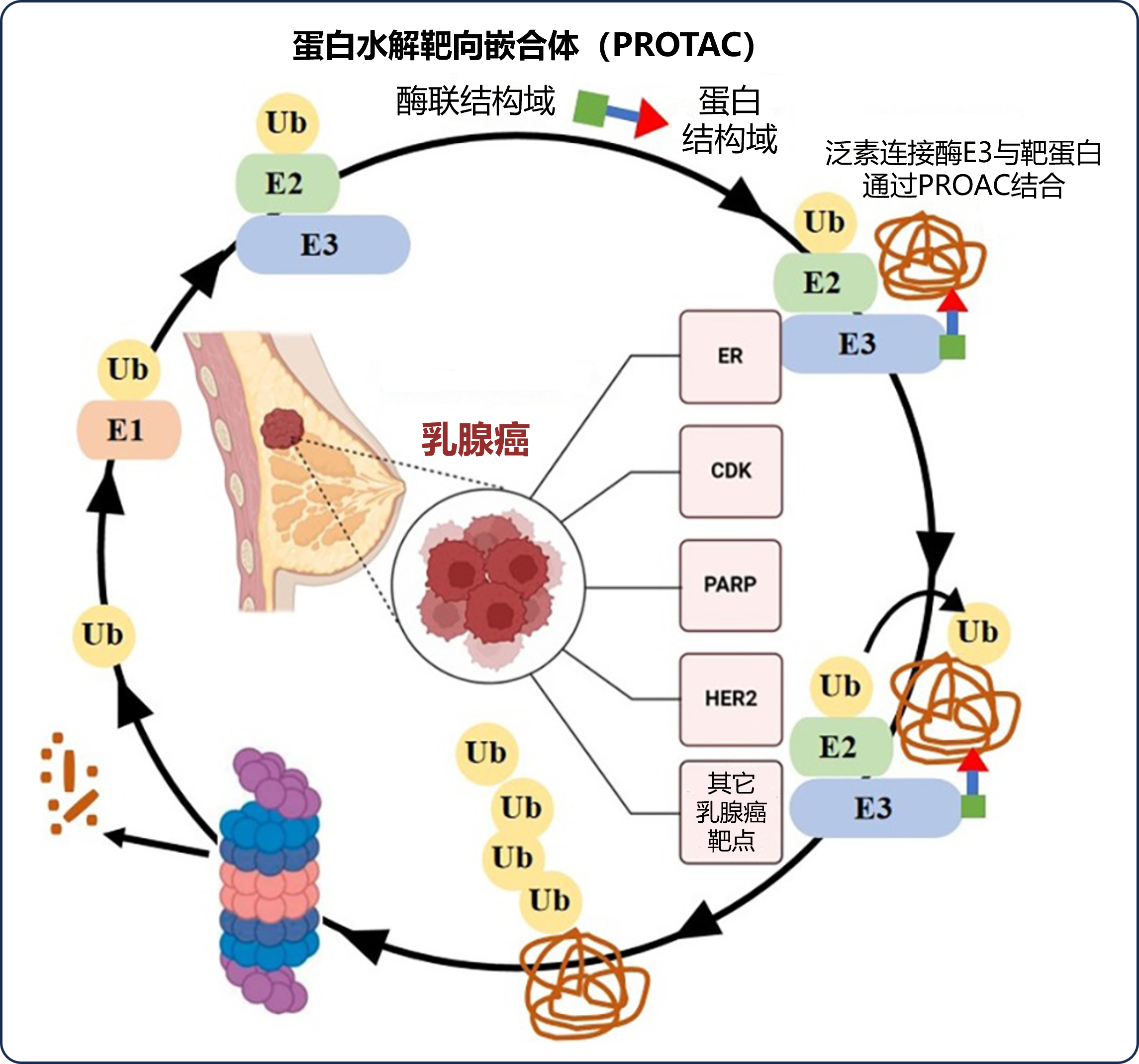
图3 蛋白水解靶向嵌合体(PROTAC)降解蛋白质的机制(在乳腺癌治疗中)
图片改编自:PROTAC: Novel degradable approach for different targets to treat breast cancer. FIG.3
Ub:蛋白质经泛素化反应修饰;E1、E2、E3:泛素连接酶E1、E2、E3;ER:雌激素受体;CDK:周期蛋白依赖性激酶;PARP:聚ADP-核糖聚合酶;HER2:人表皮生长因子受体-2
2. PROTAC等药物在肿瘤等疾病治疗中的临床前景
尽管通过小分子诱导蛋白质降解在机制上具有多样性,但临床上批准用于相关疾病治疗的药物较少。目前多种PROTAC处于不同临床试验阶段,其靶向蛋白包括雌激素受体(ER)、雄激素受体(AR)等,并涉及多个适应症,包括实体肿瘤(包括乳腺癌、前列腺癌、肺癌)、血液系统恶性肿瘤、滑膜肉瘤和自身免疫性疾病(见表1)。除PROTAC外,选择性雌激素受体降解剂(SERD)也应用了TPD技术理念而靶向降解ER,并有多种新药在研,其适应症相较PROTAC较窄,多用于乳腺癌治疗(见表1)[3]。
表1 一些应用降解剂分子类药物治疗肿瘤等疾病的临床试验

NA:不明确
E3:泛素连接酶E3;ER:雌激素受体; CRBN:泛素连接酶E3的配体(cereblon);AR:雄激素受体;IRAK4:白细胞介素-1 受体相关激酶4;IKZF1/3:Ikaros 家族锌指结构1/3; MYD88:表达TLR/MyD88/NF-κB信号通路中重要接头蛋白分子的基因;STAT3:信号传导及转录激活蛋白3;BTK:布鲁顿酪氨酸激酶;BCL-XL:B细胞淋巴瘤-2抗凋亡蛋白家族的成员蛋白;VHL:泛素连接酶E3的配体(Von Hippel-Lindau);BRAF V600:编码丝氨酸/苏氨酸蛋白激酶的基因,编码子600是其常见突变点;NTRK:神经营养性受体酪氨酸激酶,是负责编码原肌球蛋白相关激酶(Trk)的基因
二. 乳腺癌的流行病学特征、治疗方案及PROTAC的应用
1.乳腺癌的流行性病学特征及治疗方案简介
在全球范围内,乳腺癌的发病率持续上升[2]。而我国乳腺癌的流行病学形势同样不容乐观。2020年调查显示,我国乳腺癌新发超过40万例,死亡超过11万例。每年新发乳腺癌患者中约3%~10%在确诊时即有远处转移。早期患者中约有30%可发展为晚期乳腺癌,而晚期乳腺癌患者5年生存率仅为20%[4]。
从流行病学角度看,大约80%的乳腺癌患者的肿瘤依赖于ER+这一触发因素来维持其生长和生存。目前,临床上已有多种用于针对ER活性的抗乳腺癌药物,包括芳香化酶抑制剂、选择性ER调节剂(如他莫西芬)、选择性ER降解剂(如氟维司群)以及新型口服ER降解剂等。
从机制而言,他莫昔芬与ER的AF2结构域结合从而抑制基因转录,而AF1结构域介导的基因转录仍可能发生,因此他莫昔芬只能部分阻断ER的信号。氟维司群通过与ER竞争性结合,同时阻断了AF1与AF2结构域的功能,更好的抑制ER的基因转录能力;同时氟维斯群使ER的构象变得不稳定,最终导致ER被非特异性降解,ER蛋白降解率为40%-50%[5]。PROTAC的独特机制则可更彻底地阻断ER途径,通过给ER打上泛素标签,诱导ER被泛素-蛋白酶体系统特异性地降解,临床数据显示靶向ER的PROTAC药物可降低患者ER蛋白的平均降解率达71%[6]。最近的研究发现,通过口服可以降低ER水平的 PROTAC有助于有效治疗更多的ER+乳腺癌患者[2]。
2. PROTAC在乳腺癌中的应用
(1)针对ER的PROTAC
ER属核受体超家族成员,常作为配体依赖性转录因子,参与细胞核内与“激活”相关的基因转录,其常见的亚型有ERα和ERβ两种。在肿瘤疾病发展过程中,雌激素通过ER介导发挥作用,刺激乳腺肿瘤的增殖和发展[2]。
国内外多研究团队在多年研究中研发了多种针对ER的PROTAC。早在2008年,国外学者便已设计了一种由低氧诱导因子-1α(HIF-1α)作为VHL配体的ERα PROTAC,通过阻滞细胞周期中的DNA合成前期(G1期),从而抑制乳腺肿瘤细胞的ERα依赖性增殖。随着PROTAC设计及合成技术的不断成熟,PROTAC类药物已发展出越来越多的新构型,并在临床研究方面取得了多元化成果[2]。
ARV-471是第一个进入临床研发阶段的选择性靶向野生型和突变型 ER 的口服 PROTAC蛋白质降解剂,可直接结合泛素连接酶E3和ER,触发ER泛素化及随后的蛋白酶体降解。2023年,欧洲肿瘤内科学会(ESMO)年会上发布了一项1/2期,多中心,开放标签,首次人体试验(以下简称ESMO-2023)结果。该研究共纳入83例患者,旨在评估ARV-471对经治的ER+/HER2-局部晚期或转移性乳腺癌症患者的临床疗效。研究结果显示:ARV-471具有抗肿瘤活性,总体临床获益率(CBR)为36.1%(95%CI:25.9-47.4)。研究中61例患者可测量基线期疾病情况,其中7例已确认部分缓解,客观缓解率(ORR)为11.5%(95%CI:4.7-22.2)[7]。
ESMO-2023试验结果还表明,ARV-471可使ESR1(雌激素受体1基因)突变患者获益。研究结果显示:经ARV-471治疗后ESR1突变亚组患者CBR相较总体CBR更高,达48.8%(95% CI:33.3-64.5),且经ARV-471治疗1个周期后,ESR1基因突变体相关循环肿瘤基因水平显著下调,并持续多个治疗周期[7]。
除单药治疗外,ARV-471在与其它药物联合治疗中同样有显著获益。在2023年圣安东尼奥乳腺癌研讨会(SABCS)上发布了一项1b期临床研究(以下简称SABCS-2023)结果,旨在评估ARV 471+哌柏西利治疗ER+/HER2-晚期乳腺癌患者的疗效和安全性。研究结果显示,在疗效方面,总体上(n=46),经ARV 471+哌柏西利治疗后患者的中位无进展生存期为11.1月(8.2月~未达到统计学终点)。CBR为63.0%(95%CI:47.5-76.8),对其中可评估缓解的患者(n=31),ORR为41.9%(95%CI:24.5-60.9)[8]。
2023年SABCS大会报告了一项研究也表明ARV 471+哌柏西利治疗同样可使ESR1突变亚组患者获益。研究结果显示:经ARV 471+哌柏西利治疗后患者的中位无进展生存期为11.0月,(8.2月~未达到统计学终点)。且CBR和ORR指标改善情况相较总体样本改善情况更好,分别为72.4%(95%CI:52.8–87.3)和47.1%(95%CI:23.0-72.2)。此外,在安全性方面,SABCS-2023研究显示ARV 471+哌柏西利治疗中无5级不良事件和剂量限制性毒性,也无发热性中性粒细胞减少相关不良事件[8]。
(2)针对CDK的PROTAC
CDK属丝氨酸/苏氨酸蛋白激酶家族成员,参与协调细胞周期,是保障细胞正常分裂和增殖的重要环节。其中CDK4/6是细胞进入DNA合成期的关键因子,在肿瘤的发生和发展中发挥重要作用[2]。
2020年,国外学者以CRBN、VHL等为配体,基于哌柏西利设计研发了一系列针对CDK4/6的PROTAC,并证实了其在Jurkat 细胞(来源于人类的T细胞白血病细胞系)中对CDK4/6的破坏作用。同年,其他国外学者通过筛选不同的泛素连接酶E3,并通过不同的连接子将其与哌柏西利偶联,进而对CDK4/6 PROTAC的性质进行了全面分析,并将CDK6 PROTAC的特异性谱系扩展到VHL(在大多数恶性肿瘤细胞中很少失活的关键分子)等配体库。2023年,我国学者应用不同的连接子,将哌柏西利与KB02(一种泛素连接酶E3配体)结合,研究发现其中CDK4/6复合物表现显著的抑制活性,并可特异性下调MDA-MB-231细胞(一种人乳腺癌细胞)中的CDK4/6浓度并延长细胞周期。除CDK4/6外,有关CDK12/13 PROTAC在临床应用方面也取得了诸多成果[2]。
(3)针对PARP的PROTAC
聚ADP-核糖聚合酶(PARP)是DNA修复和保障DNA完整性的关键酶,其家族包含17个成员,其中PARP-1占PARP活性的90%以上,是基因损伤修复的重要决定因素,而PARP-2功能与PARP-1相似,但在底物选择上不同[2]。
在机制上,传统PARP靶向抑制剂可通过与肿瘤细胞中PARP-1及PARP-2结合,阻止其脱离DNA损伤位点,导致DNA复制受阻和同源重组缺陷,进而加速肿瘤细胞DNA双链断裂和死亡,但这一过程仍然存在耐药性的问题。而PROTAC作用机制在于降解致病靶蛋白,对克服耐药性方面具有很大优势。早在2019年,我国学者就已设计出针对PARP-1的PROTAC,并在MDA-MB-231细胞中观察到PARP-1的降解和细胞凋亡。此后,我国学者又于2022年构建了以PARP-1抑制剂奥拉帕尼为配体的PROTAC(NN3),其通过减少诱导铁死亡途径,在p53阳性乳腺癌细胞中表现出独特的抗肿瘤作用。在国外,多方学者也应用不同配体,针对PARP研发了多种具有抗肿瘤作用的PROTAC[2]。
总结
随着科学技术的发展与进步,抗肿瘤治疗也在不断地突破创新。PROTAC等药物运用TPD新兴手段,打破了传统的占位驱动型抑制剂对缺乏催化活性口袋的蛋白质难以靶向结合的限制,且PROTAC通过UPS介导致病靶蛋白降解发挥独特的抗肿瘤作用,一定程度上克服了靶向治疗的耐药性。目前,乳腺癌在全球及我国范围内流行病学形势仍不容乐观,而PROTAC的出现无疑给乳腺癌治疗,尤其是ER+患者的治疗指明新的方向。在具体临床应用方面,国内外学者利用多种配体,研发出针对ER、CDK及PARP等乳腺癌相关靶蛋白的PROTAC,并证实了这些药物在抗乳腺肿瘤方面的积极作用。可以预见,在不久的将来,PROTAC会成为临床医生对抗乳腺肿瘤的又一有利武器。
参考文献
- Fang Y, Wang S, Han S, et al. Targeted protein degrader development for cancer: advances, challenges, and opportunities. Trends Pharmacol Sci. 2023 May;44(5):303-317.
- Wang Z, Che S, Yu Z. PROTAC: Novel degradable approach for different targets to treat breast cancer. Eur J Pharm Sci. 2024 May 11;198:106793.
- Tsai JM, Nowak RP, Ebert BL, et al. Targeted protein degradation: from mechanisms to clinic. Nat Rev Mol Cell Biol. 2024 Apr 29.
- 国家肿瘤质控中心乳腺癌专家委员会, 中国抗癌协会乳腺癌专业委员会, 中国抗癌协会肿瘤药物临床研究专业委员会. 中国晚期乳腺癌规范诊疗指南(2022版)[J].中华肿瘤杂志, 2022, 44(12): 1262-1287.点击查看
- Kuter I, Gee JM, Hegg R, et al. Dose-dependent change in biomarkers during neoadjuvant endocrine therapy with fulvestrant: results from NEWEST, a randomized Phase II study. Breast Cancer Res Treat. 2012 May;133(1):237-46.
- Sara A Hurvitz, Anne F Schott, Cynthia Ma, et al. ARV-471, a PROTAC® estrogen receptor (ER) degrader in advanced ER-positive/human epidermal growth factor receptor 2 (HER2)-negative breast cancer: phase 2 expansion (VERITAC) of a phase 1/2 study. SABCS, 2023, GS3-03.
- Hamilton E P, Han H S, Schott A F, et al. Vepdegestrant, a PROteolysis TArgeting Chimera (PROTAC) Estrogen Receptor Degrader, in Estrogen Receptor+/Human Epidermal Growth Factor Receptor 2- Advanced Breast Cancer: Update of Dose Escalation Results From a Phase 1/2 Trial. ESMO, 2023, Poster 390P.
- Hamilton E P, Jeselsohn R, Hurvitz S A. Vepdegestrant, a PROteolysis TArgeting Chimera (PROTAC) Estrogen Receptor Degrader, Plus Palbociclib in Estrogen Receptor–Positive/Human Epidermal Growth Factor Receptor 2–Negative Advanced Breast Cancer: Phase 1b Cohort. SABCS, 2023, PS15-03.



