本品适用于成人偏头痛的急性治疗(有或无先兆)。1
一般人群用法用量
(1)用量1
- 本品推荐剂量为每次口服75 mg,按需服药。每日不超过一次。
- 尚未确定在30天内使用超过18次的安全性。
(2)用法1,2
- 本品推荐剂量为每次口服75 mg,按需服药。每日不超过一次。
- 图片说明:
 |
|
 |
|
- 视频说明:
肾功能损害人群用法用量
- 轻度、中度或重度肾功能损害患者无需调整用药剂量。1
- 重度肾功能损害患者频繁使用本品时应谨慎。1
- 终末期肾病(肌酐清除率[CLcr]<15 mL/min)患者应避免使用瑞美吉泮。1
- 一项临床研究中,分别将轻度(CLcr 60-89 mL/min)、中度(CLcr 30-59 mL/min)和重度(CLcr 15-29 mL/min)肾功能损害受试者与汇总的健康对照受试者中瑞美吉泮的药代动力学数据进行了比较,结果显示75 mg单次给药后,在轻、中度肾功能损害受试者中,总瑞美吉泮的暴露量(血浆浓度时间曲线下面积[AUC])增加不到50%。在重度肾功能损害受试者中,未结合瑞美吉泮的暴露量(AUC)是健康受试者的2.57倍。尚未在终末期肾病(CLcr<15 mL/min)患者中进行研究。1
肝功能损害人群用法用量
- 轻度(Child-Pugh A)或中度(Child-Pugh B)肝功能损害患者无需调整剂量。1
- 重度(Child-Pugh C)肝功能损害患者应避免使用瑞美吉泮。重度肝功能损害受试者的瑞美吉泮血浆浓度(未结合瑞美吉泮的血浆浓度时间曲线下面积[AUC])显著增高。1
- 一项临床研究比较了轻度、中度和重度肝功能损害受试者与各组相匹配的健康对照受试者中瑞美吉泮的药代动力学数据,结果表明与肝功能正常的健康受试者相比,重度肝功能损害受试者接受瑞美吉泮75 mg单次给药后的暴露量(未结合AUC)是健康受试者的3.89倍。1
本品用于偏头痛急性治疗时最常见的不良反应为恶心(1.2%),多数严重程度为轻或中度。接受治疗的患者中超敏反应(包括呼吸困难和严重皮疹)的发生率低于1%。1
下表按系统器官分类列出的不良反应,对每种药物不良反应的发生率进行分类:十分常见(≥1/10);常见(≥1/100 至<1/10);偶见(≥1/1,000 至<1/100);罕见(≥1/10,000 至<1/1,000);十分罕见(<1/10,000)。
表1 不良反应种类和频率1
| 系统器官分类 | 不良反应 | 发生率 |
|---|---|---|
| 免疫系统疾病 | 超敏反应,包括呼吸困难和严重皮疹 | 偶见 |
| 胃肠系统疾病 | 恶心 | 常见 |
在临床研究接受治疗的患者中,超敏反应(包括呼吸困难和皮疹)的发生率低于1%,超敏反应(包括严重超敏反应)可发生在给药后数天,也发生过迟发型严重超敏反应。如果发生超敏反应,应停用瑞美吉泮并给予适当治疗。1
在两项为期1年、开放标签的延伸试验中评估了瑞美吉泮的长期安全性;1662例患者接受了至少6个月的瑞美吉泮治疗,740例患者接受了12个月的瑞美吉泮治疗。1
药物相互作用:1
- 不建议将瑞美吉泮与强效CYP3A4抑制剂(克拉霉素、伊曲康唑、利托那韦)合并使用。
- 不建议与强效CYP3A4诱导剂(苯巴比妥、利福平、圣约翰草 [贯叶连翘])或中效CYP3A4诱导剂(波生坦、依非韦伦、莫达非尼)合并使用。
- 如果与CYP3A4中效抑制剂(地尔硫卓、红霉素、氟康唑)或P-gp 强效抑制剂(环孢素、维拉帕米、奎尼丁)合并使用时,应避免在48小时内再次给予瑞美吉泮。
药物过量性头痛:1
- 过量使用任何类型的药物治疗头痛均可导致其恶化。
- 对于频繁出现或每日发作的头痛,即使已经常规使用了药物治疗急性头痛,或由于常规使用药物导致了头痛,均应考虑药物过量性头痛(MOH)的诊断。
- 如果已出现或怀疑出现这种情况,应立即就医,并停止治疗。
妊娠期:1
- 妊娠妇女服用瑞美吉泮的数据有限。
- 作为预防措施,建议在妊娠期间避免使用本品。
- 动物研究表明,瑞美吉泮没有胚胎致死作用,且在临床相关暴露量下未观察到致畸风险。在妊娠期间给予瑞美吉泮后,仅在与母体毒性相关的暴露水平(约为临床暴露量的200 倍)下观察到对胚胎-胎仔发育的不良影响(大鼠胎仔体重下降和骨骼变异增加)。
- 妊娠大鼠于器官发生期经口给予硫酸瑞美吉泮10、60、300 mg/kg/天,高剂量可导致与母体毒性相关的胎仔体重降低和胎仔骨骼变异发生率增加。大鼠胚胎发育毒性无影响剂量(60 mg/kg)下的血浆暴露量约为人最大推荐剂量(MRHD)暴露量的45倍。妊娠兔于器官发生期经口给予硫酸瑞美吉泮10、25、50 mg/kg/天,未见对兔胚胎发育的不良影响,高剂量(50 mg/kg/天)下兔血浆暴露量约为人MRHD暴露量的10倍。大鼠于妊娠期和哺乳期经口给予硫酸瑞美吉泮10、25、60 mg/kg/天,对大鼠围产期发育未见影响,高剂量(60 mg/kg/天)下大鼠血浆暴露量约为人MRHD暴露量的24倍。
哺乳期:1
- 目前尚没有关于母乳分泌量是否会受到影响的数据,临床使用时应考虑母乳喂养对发育和健康的获益,以及母亲对本品的临床需求,以及瑞美吉泮或母体基础疾病对母乳喂养婴儿的任何潜在不良反应。
- 在一项单中心研究中,12名哺乳期女性接受了瑞美吉泮75 mg单次给药,在她们的乳汁中仅检出极低浓度的瑞美吉泮。相对婴儿剂量(RID)不足1%。
生育能力:1
- 动物研究显示本品对雌性和雄性生育能力无临床相关的影响。
- 雄性和雌性大鼠在交配前和交配期间,雌性大鼠持续给药至妊娠期第7天经口给予硫酸瑞美吉泮30、60、150 mg/kg/天,高剂量可导致生育力降低。降低剂量进行第二项试验,给药剂量为5、15、25 mg/kg/天,未见对生育力和早期胚胎发育的不良影响。大鼠生育力和早期胚胎发育毒性无影响剂量(60 mg/kg/天)下的血浆暴露量约为人最大推荐剂量(MRHD)75 mg/天暴露量的30倍。
活性成分:硫酸瑞美吉泮
辅料:明胶,甘露醇,三氯蔗糖,薄荷香精
48个月
本品是一种降钙素基因相关肽(CGRP)受体拮抗剂。1
表2 关键临床概览

MBS:最困扰症状,定义为恶心、畏声或畏光
研究概述:1,3
- 这是一项在中国和韩国开展的、双盲、随机、多中心、评估瑞美吉泮相比安慰剂治疗中度或重度偏头痛的安全性和有效性的研究。
- 受试者被分发一剂试验用药品,即瑞美吉泮75 mg或匹配的安慰剂。
- 研究的总持续时间约为11周。包括3-28天的筛选期、急性治疗期以及在试验用药品给药后7天内的治疗结束(EOT)访视。
- 研究随机入组了1431例受试者。
关键纳入标准:3
- 在入组前3个月内:
- 每月中重度偏头痛发作≥2次
- 每月头痛天数<15天
关键排除标准:3
- 目前有证据表明有未控制、不稳定或最近确诊的心血管疾病
研究终点:1,3
- 主要终点:给药后2小时无痛比例、给药后2小时无最困扰的症状 (MBS) 比例。
- 次要终点:给药后2小时的疼痛缓解率、给药后2小时功能恢复正常的比例、给药后24小时内的补救药物使用情况、给药后2-24小时的持续无疼痛率、给药后2-48小时的持续无疼痛率。
单剂给药后2小时患者无痛比例,瑞美吉泮组显著优于安慰剂组。3
图1 2小时无痛患者比例3

单剂给药后2小时患者无最困扰症状(MBS)比例,瑞美吉泮组显著优于安慰剂组。3
图2 2小时无MBS患者比例3

MBS:最困扰症状
单剂给药后45分钟患者疼痛缓解比例,瑞美吉泮组显著优于安慰剂组,45分钟至3小时患者疼痛缓解比例,瑞美吉泮组持续显著优于安慰剂组。3
图3 单剂给药后45分钟至3小时患者疼痛缓解比例,瑞美吉泮组显著优于安慰剂组。3

瑞美吉泮组在给药后90分钟的无痛方面的疗效显著优于安慰剂组。3
图4 用药后不同时间无痛患者比例3

瑞美吉泮组在给药后45分钟的无最困扰症状(MBS)方面的疗效显著优于安慰剂组。3
图5 用药后不同时间无MBS患者比例3

单剂给药后60分钟患者恢复正常功能比例,瑞美吉泮组显著优于安慰剂组,60分钟至8小时患者恢复正常功能比例,瑞美吉泮组持续显著优于安慰剂组。3
图6 单剂给药后60分钟至8小时功能障碍恢复正常的患者比例,瑞美吉泮组显著优于安慰剂组。3

单剂给药后2小时患者疼痛缓解比例,瑞美吉泮组显著优于安慰剂组。3
图7 2小时疼痛缓解3

单剂给药后2小时患者功能障碍恢复正常比例,瑞美吉泮组显著优于安慰剂组。3
图8 2小时功能障碍恢复正常3

单剂给药后24小时内患者使用补救药物比例,瑞美吉泮组显著优于安慰剂组。3
图9 24小时内使用补救药物3

单剂给药后患者2-24小时和2-48小时持续无疼痛率,瑞美吉泮组显著优于安慰剂组。3
图10 2-24小时和2-48小时持续无疼痛率3

- 中韩3期研究中国患者占比>80%。研究整体的安全性能够体现中国患者服用瑞美吉泮后的安全性。3
- 瑞美吉泮组和安慰剂组不良反应总体发生率相当(16% vs17%),大多数不良事件为轻度或中度,TEAEs、严重TEAEs以及药物相关的严重TEAEs,安慰剂组的发生率均高于或等于瑞美吉泮组,且瑞美吉泮组没有任何患者出现与药物相关的严重不良反应。3
表3 中韩3期研究不良事件概览

AE=不良事件;TEAE=治疗期间不良事件。
基于瑞美吉泮中韩3期临床试验的研究结果,对1075名中国受试者进行子集分析:6
- 瑞美吉泮75 mg用于中国成人偏头痛的急性治疗有效,具有快速、持续的疗效,安全性和耐受性和安慰剂组相当。
- 这些结果证实了先前研究的发现,并且与总体试验人群的结果相似。单剂给药后患者2小时无痛和无最困扰症状(MBS)比例,瑞美吉泮组显著优于安慰剂。
图11 中国患者亚组分析共同主要终点 瑞美吉泮在改善服药后2小时无疼痛及无MBS患者比例

MBS:最困扰症状
图12 中国患者亚组分析关键次要终点 瑞美吉泮所有的关键次要终点改善均显著优于安慰剂6
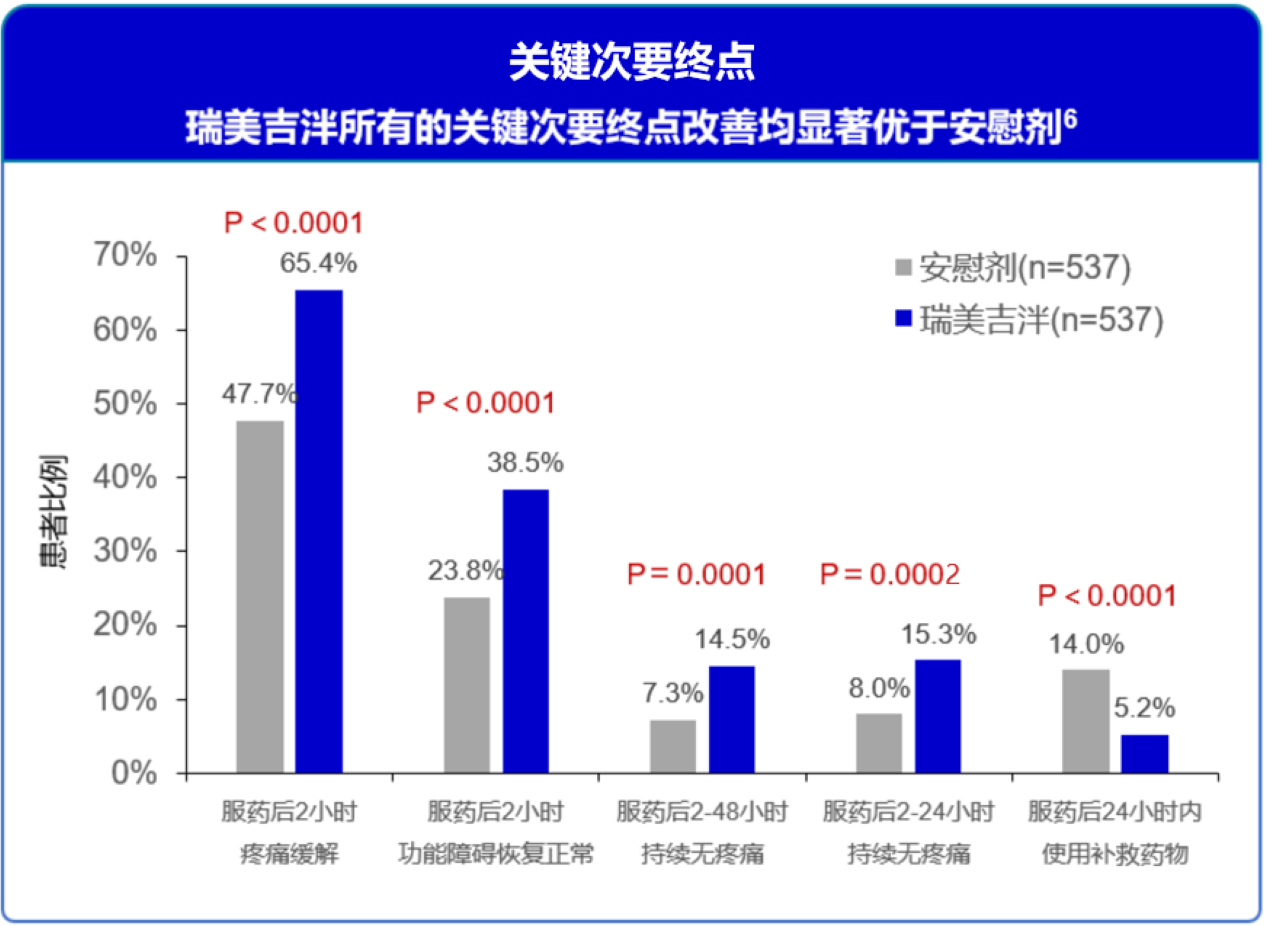
基于瑞美吉泮中韩3期临床试验的研究结果,对1075名中国受试者进行子集分析:6
- 瑞美吉泮的总体安全性与安慰剂相当,因此在中国受试者身上观察到的安全性结果也与之前在美国进行的瑞美吉泮试验结果一致。
- 在中国受试者亚组中,瑞美吉泮组未出现与治疗相关的严重TEAE,大多数TEAE的严重程度为轻度或中度,大多数TEAE无需治疗即可缓解,TEAE在瑞美吉泮组和安慰剂组中的发生率相似,未观察到心电图或常规实验室检查有临床意义的变化,也未出现肝毒性信号。
表4 所有接受治疗的中国受试者的治疗期间不良事件概要
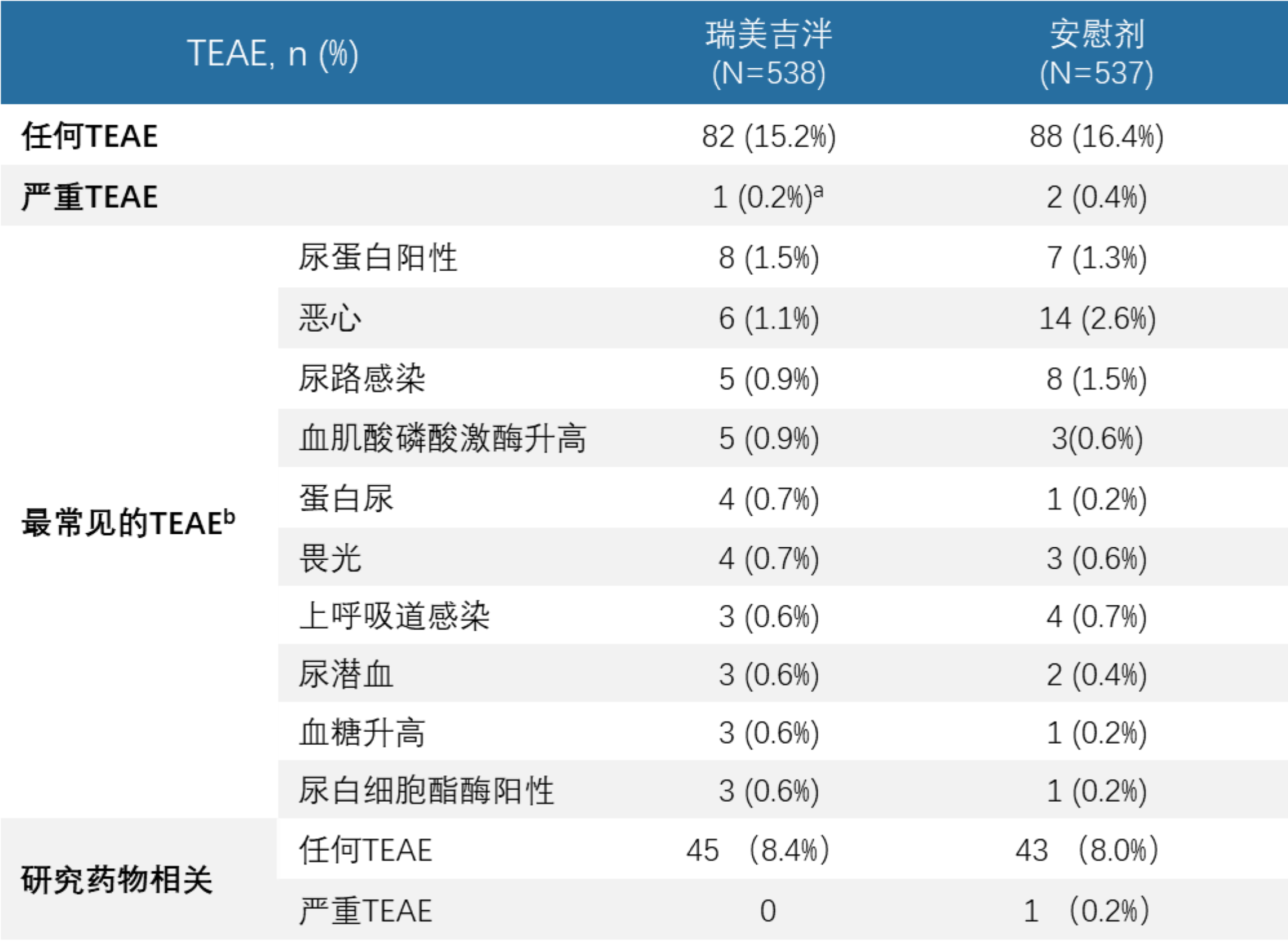
TEAE=治疗期间不良事件
a该严重TEAE使用MedDRA(23.0版)首选术语“感染”进行分类。
b瑞美吉泮口崩片75 mg组有≥0.5%的受试者出现TEAE。
无论患者是否同时使用预防性药物,瑞美吉泮均可显著提高无痛患者比例。
瑞美吉泮中韩3期研究中,98例(7.3%)受试者既往使用过预防性偏头痛药物*,使用最多的前三种预防性药物分别为:氟桂利嗪,阿米替林,托吡酯。7
表5 瑞美吉泮中韩3期亚组分析

风险差异 (95%CI)
*允许使用预防性偏头痛药物的受试者继续接受该治疗,只要这些受试者在筛选访视前接受稳定剂量治疗至少已达3个月,且该剂量预计不会在研究期间改变。
MBS:最困扰症状
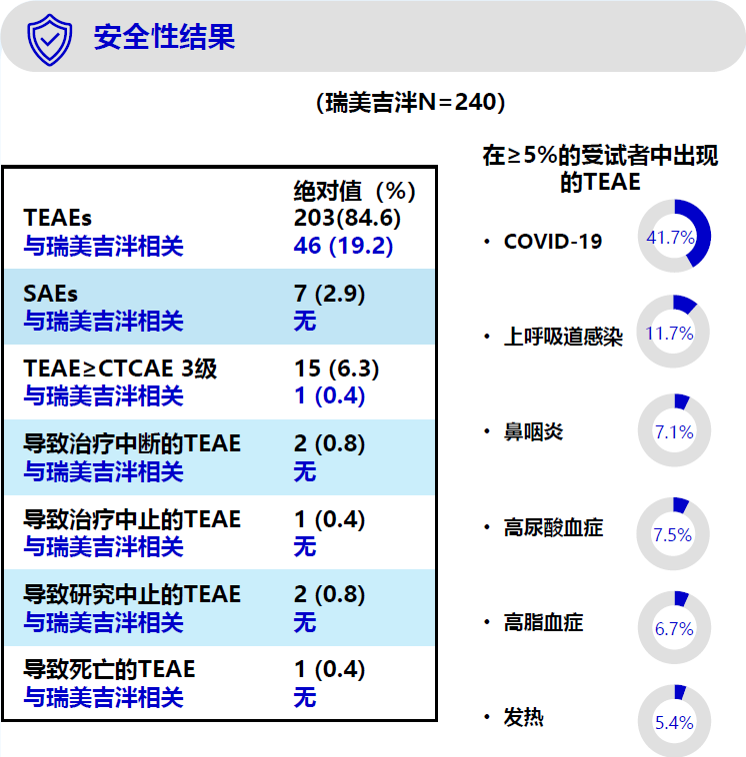
- 本研究证实了中国偏头痛患者使用瑞美吉泮急性治疗1年,其安全性和耐受性良好。
- 瑞美吉泮75 mg 按需使用治疗偏头痛急性发作,从第一个月起即减少患者每月偏头痛天数(MMDs)和疼痛强度。治疗一个月后,MMDs平均值从11.2天(基线)降低至9.5天。治疗11个月后,MMDs进一步降低至5.5天。
- 患者每月服用瑞美吉泮的片数逐渐下降
- 使用瑞美吉泮按需治疗1年,偏头痛专用生活质量调查问卷(MSQoLQ)问卷评分(限制性角色功能、预防性角色功能,情感功能)均显著改善(52.7→77、62.3→85、63.2→86.2);偏头痛失能评估测试问卷(MIDAS)评分显著改善(55.1→ 17.3)。
- 本研究证实,瑞美吉泮按需治疗可以持续降低偏头痛发作频率,有效阻止发作性偏头痛向慢性偏头痛的转化
研究概述:4
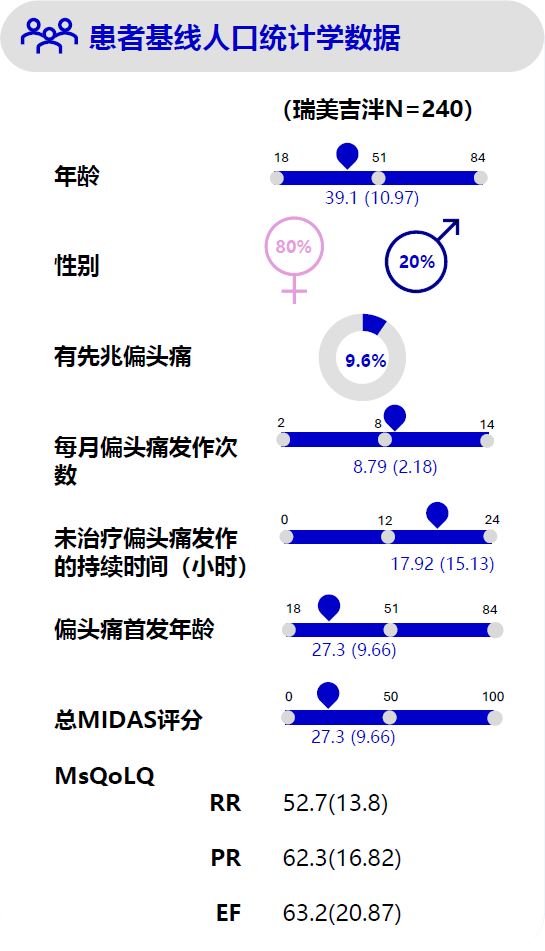
本研究是一项多中心、开放研究,目的是在中国偏头痛受试者中评估瑞美吉泮75 mg长期给药(按需给药,每个日历日最多给药1片)的安全性和耐受性。研究入组240名受试者,筛选期为30天,疗程期限为52周±7天,随后进行14±2天的随访。
纳入标准和筛选过程:4
纳入标准包括年龄≥18岁,至少有1年的偏头痛史(根据国际头痛分类第3版[beta版]),首次偏头痛发作年龄小于50岁,偏头痛发作持续时间为4-72小时(如果未接受治疗),以及在30天的观察期内至少有6天的偏头痛发作。此外,筛选前3个月内,每月中重度偏头痛发作次数应在6-18次之间。该研究允许使用预防性偏头痛药物的患者也可入组。
研究终点:4
研究的主要终点包括不良事件的记录,包括常见不良事件(发生率≥5%)、严重不良事件、导致停药的不良事件以及检查数据等。次要终点包括每4周偏头痛发作的天数(与观察期相比)、每4周偏头痛发作的严重程度(与观察期相比)。此外,还设有探索性终点,包括偏头痛-特异生活质量问卷(MSQoLQ)评估的生活质量变化、偏头痛用药偏好问卷(PoM)结果变化、药物满意度问卷结果变化、偏头痛残疾程度评估测试(MIDAS)结果变化以及临床总体印象量表(CGI-c)的变化评分。
头痛发作频率
对于所有严重程度(轻中重度)的偏头痛发作
- 按需服用瑞美吉泮一个月MMDs平均降低1.7天,MMDs由基线11.2天降至9.5天
- 至第11个月MMDs降低5.7天,MMDs降至5.5天
用药片数
患者每月服用瑞美吉泮的片数逐渐下降
肝功能异常(2.5%)
共有6例患者出现肝功能异常且被研究者判断为与药物相关
- 其中5例因AST/ALT变化程度轻微,被研究者判定为CTCAE 1-2级
- 1例被判定为CTCAE3级(中度毒副反应,肝功能出现轻度异常、食欲下降等,会对日常生活和工作造成一定影响,需要医生干预治疗,可能需要使用抗恶心药物、止吐药物)
- 该受试者入组时肝功能正常。服药期间受试者曾剧烈运动和饮酒
- 服药第8周:ALT 112U/L(2.2 × ULN,标准值:9-50 U/L),AST 433 U/L(10.8 × ULN,标准值:15-40 U/L)
- 研究者评估后未做干预。15天后ALT,AST恢复正常
- 研究者将该事件评估为可能与药物有关,事件结局评估为痊愈。外部肝脏专家小组对该病例进行了审查,结论是:TEAE与运动诱导的肌肉损伤表现完全一致,无药物性肝损伤的证据。申办者评估事件为与药物无关
实验室检查异常
| 瑞美吉泮(N=240) | CTCAE分级 绝对值(%) | |
|---|---|---|
| 1-2级 | 3-4级 | |
| 高甘油三酯血症 | 50(20.8%) | 2(0.8%) |
| 淋巴细胞计数减少 | 36(15.0%) | 1(0.4%) |
| 贫血 | 34(14.2%) | 1(0.4%) |
| 中性粒细胞减少 | 33(13.8%) | 1(0.4%) |
| 白细胞减少 | 29(12.1%) | 1(0.4%) |
| AST升高 | 27(11.3%) | 1(0.4%) |
| CPK升高 | 10(4.2%) | 4(1.7%) |
| 低钾血症 | 2(0.8%) | 1(0.4%) |
研究概述:1,5
- 这是一项在美国开展的、双盲、随机、多中心、评估瑞美吉泮相比安慰剂治疗中度或重度偏头痛的安全性和有效性的研究。
- 受试者被分发一剂试验用药品,即瑞美吉泮75mg或匹配的安慰剂。
- 研究的总持续时间约为11周。包括3-28天的筛选期、急性治疗期以及在试验用药品给药后7天内的治疗结束(EOT)访视。
- 研究随机入组了1811例受试者。
关键纳入标准:5
- 在入组前3个月内:
- 每月中重度偏头痛发作2-8次
- 每月头痛天数<15天
关键排除标准:5
- 目前有证据表明有未控制、不稳定或最近确诊的心血管疾病
研究终点:1,5
- 主要终点:给药后2小时无痛比例、给药后2小时无最困扰的症状 (MBS) 比例。
- 次要终点:给药后2小时的疼痛缓解率、给药后2小时功能恢复正常的比例、给药后24小时内的补救药物使用情况、给药后2-24小时的持续无疼痛率、给药后2-48小时的持续无疼痛率。
瑞美吉泮组比安慰剂组在服药后2小时无疼痛及无最困扰症状(MBS)的患者比例更高。5
图18 主要终点:给药后2小时无痛、无MBS患者比例

MBS=最困扰症状
瑞美吉泮组和安慰剂组分别有90名(13%)和73名(11%)患者报告不良事件(AE)。最常见的AE为恶心(瑞美吉泮2%,安慰剂1%)和尿路感染(瑞美吉泮1%,安慰剂1%)。两组患者均未报告严重不良事件。5
无先兆偏头痛诊断标准(ICHD-3)

国际头痛疾病分类(ICHD),由国际头痛协会(IHS)发布,目前为2018年出版的第三版ICHD-38
有先兆偏头痛诊断标准(ICHD-3)

国际头痛疾病分类(ICHD),由国际头痛协会(IHS)发布,目前为2018年出版的第三版ICHD-38
发作性偏头痛和慢性偏头痛诊断标准(ICHD-3)

a 不符合慢性偏头痛标准的人
b 每年有2.5%的发作偏头痛患者发展为慢性偏头痛
ICHD-3:国际头痛疾病分类,第3版;MHD:每月头痛天数;MMD:每月偏头痛天数





- 月经性偏头痛诊断和治疗中国专家共识(2025版)
- 2024 月经性偏头痛诊断与治疗中国专家共识
- 2024 中国偏头痛急性期治疗指南(第一版)
- 2024 IHS全球实践建议:偏头痛的急性药物治疗
- 成人原发性头痛的规范化管理:泛长三角头痛诊疗专家建议(2024)
- 2024 中国药物过度使用性头痛诊治指南(第一版)
- 2024 法国头痛协会(SFEMC)意见书:偏头痛治疗
- 2023 中国 偏头痛诊断与治疗指南
- 2023 英国 偏头痛药物治疗指南
- 2023 NICE 瑞美吉泮用于成人偏头痛急性治疗
- 2022 中国 偏头痛诊治指南
- 2021 美国 头痛协会共识声明:将新的偏头痛治疗药物纳入临床实践的最新信息
- 2021 法国 成人偏头痛的诊断和治疗指南-药物治疗
- 2021 法国 成人偏头痛的诊断和治疗指南-诊断与评估
请注意:我们不建议超出国家药品监督管理局批准的药品说明书的应用或未在中国(不含港澳台)上市药品的应用。如我们所提供的文献信息及指南共识涉及该等内容,系出于传递科学知识的需要,不应视为对未在中国(不含港澳台)上市药品或已在中国上市但超出国家药品监督管理局批准的药品说明书的药品适应症/用法的推广。
- 乐泰可(硫酸瑞美吉泮口崩片)说明书
- Vydura (rimegepant) Package Insert. Pfizer Europe Inc.
- Yu S, Kim BK, Guo A, et al. Safety and efficacy of rimegepant orally disintegrating tablet for the acute treatment of migraine in China and South Korea: a phase 3, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2023 Jun;22(6):476-484.
- Yu S, Ma L, Zhong Q, et al. Interim results of a long-term safety study of 75 mg rimegepant administered as needed in acute treatment of migraine among Chinese adults. Poster session presented at: Inernational Headache Congress; 2023 Sept 14-17; Seoul, South Korea.
- Croop R, Goadsby PJ, Stock DA, et al. Efficacy, safety, and tolerability of rimegepant orally disintegrating tablet for the acute treatment of migraine: a randomised, phase 3, double-blind, placebo-controlled trial. Lancet. 2019 Aug;31;394(10200):737-745.
- Yu S, Guo A, Wang Z, et al. Rimegepant orally disintegrating tablet 75 mg for acute treatment of migraine in adults from China: a subgroup analysis of a double-blind, randomized, placebo-controlled, phase 3 clinical trial. J Headache Pain. 2024 Apr 16;25(1):57
- Yu S, Lu Z, Liu Y, et al. Efficacy of rimegepant for the acute treatment of migraine in Chinese and Korean adults receiving concurrent preventive medication. Poster session presented at: Inernational Headache Congress; 2023 Sept 14-17; Seoul, South Korea.
- Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd edition. Cephalalgia. 2018;38(1):1-211.
- Katsarava Z, Buse DC, Manack AN, et al. Defining the differences between episodic migraine and chronic migraine. Curr Pain Headache Rep. 2012 Feb;16(1):86-92.
- Buse DC, Manack AN, Fanning KM, et al. Chronic migraine prevalence, disability, and sociodemographic factors: results from the American Migraine Prevalence and Prevention Study. Headache. 2012 Nov-Dec;52(10):1456-70.
- Bigal ME, Lipton RB. Modifiable risk factors for migraine progression. Headache. 2006 Oct;46(9):1334-43.
- Dodick DW, Loder EW, Manack Adams A, et al. Assessing barriers to chronic migraine consultation, diagnosis, and treatment: results from the chronic migraine epidemiology and outcomes (CaMEO) study. Headache. 2016 May;56(5):821-834.
- 郭政, 等. 疼痛诊疗学第4版. 人民卫生出版社. 2016.
- 冷希圣,韦军民,刘连新, 等. 普通外科围手术期疼痛处理专家共识[J]. 中华普通外科杂志, 2015,30(2):166-173.
- Eigenbrodt AK, Ashina H, Khan S, et al. Diagnosis and management of migraine in ten steps. Nat Rev Neurol. 2021 Aug;17(8):501-514.
- Rapoport AM, Bigal ME. ID-migraine. Neurol Sci. 2004 Oct;25 Suppl 3:S258-60.
- Láinez MJ, Domínguez M, Rejas J, et al. Development and validation of the Migraine Screen Questionnaire (MS-Q). Headache. 2005 Nov-Dec;45(10):1328-38.



